自 2000 年首次发布以来,人类参考基因组仅涵盖基因组的常染色质部分,而重要的异染色质区域尚未完成。
2022年3月31日,端粒到端粒 (T2T) 联盟在Science 在线发表题为“The complete sequence of a human genome”的研究论文(该文章入选为Science封面文章),该研究针对剩余 8% 的基因组,提供了人类基因组的完整 30.55 亿碱基对序列 T2T-CHM13,其中包括除 Y 之外的所有染色体的无间隙装配,纠正了之前的参考序列,并介绍了近 2 亿个碱基对序列,其中包含 1956 个基因预测,其中 99 个预测为蛋白质编码。完成的区域包括所有着丝粒卫星阵列、最近的节段重复和所有五个近端着丝粒染色体的短臂。
总之,与过去 20 年的任何基因组参考版本相比,T2T-CHM13 组装增加了五个完整的染色体臂和更多的额外序列。这 8% 的基因组并没有因为缺乏重要性而被忽视,而是因为技术限制。高精度长读长测序终于消除了这一技术障碍,能够对整个人类基因组的基因组变异进行全面研究,预计这将推动人类基因组健康和疾病的未来发现。
另外,2022年4月1日,约翰霍普金斯大学Sergey Aganezov等人在Science 在线发表题为“A complete reference genome improves analysis of human genetic variation”的研究论文,Telomere-to-Telomere CHM13 基因组增加了近 2 亿个碱基对序列,纠正了数千个结构错误,并为临床和功能研究解锁了人类基因组中最复杂的区域。该研究展示了该参考序列如何普遍改进读取映射和变体调用。 该研究在以前未解决的区域中识别出每个样本数十万个变体,展示了 T2T-CHM13 参考对进化和生物医学发现的重要作用。同时,该参考序列消除了每个样本数以万计的虚假变异,包括将 269 个医学相关基因中的假阳性减少多达 12 倍。由于变异发现的这些改进以及种群和功能基因组资源,T2T-CHM13 将取代 GRCh38 作为人类遗传学的主要参考。
2022年4月1日,华盛顿大学Mitchell R. Vollger等人在Science 在线发表题为“Segmental duplications and their variation in a complete human genome”的研究论文,该研究使用完整的端粒到端粒人类基因组 (T2T-CHM13),提供了人类片段重复 (SD)组织的全面视图。SD 占额外序列的近三分之一,将全基因组估计从 5.4% 增加到 7.0% [2.18 亿碱基对 (Mbp)]。对 268 个人类基因组的分析表明,91% 的先前未解析的 T2T-CHM13 SD 序列 (68.3 Mbp) 更好地代表了人类拷贝数变异。比较来自人类 (n = 12) 和非人类灵长类动物 (n = 5) 基因组的长读长组装,该研究系统地重建了生物医学相关和重复基因的进化和结构单倍型多样性。该分析揭示了人类与其他灵长类动物之间 SD 组织结构杂合性和进化差异的模式。
2022年4月1日,加州大学伯克利分校Nicolas Altemose等人在Science 在线发表题为“Complete genomic and epigenetic maps of human centromeres”的研究论文,一个完整的端粒到端粒人类基因组组装 (T2T-CHM13) 使研究人员能够全面地表征构成基因组 6.2% 的着丝粒周围和着丝粒重复序列(189.9 兆碱基)。这些区域的详细图谱显示了多碱基结构重排,包括活性着丝粒重复阵列。对着丝粒相关序列的分析揭示了着丝粒的位置与周围 DNA 通过分层重复扩展的进化之间的密切关系。此外,对不同个体组中 X 染色体着丝粒的比较揭示了这些复杂且快速进化的区域中高度的结构、表观遗传和序列变异。
2022年4月1日,康涅狄格大学Savannah J. Hoyt等人在Science 在线发表题为“From telomere to telomere: The transcriptional and epigenetic state of human repeat elements”的研究论文,该研究提出了 T2T-CHM13 人类参考基因组的从头重复发现和注释。该研究确定了以前未知的卫星阵列,扩展了重复和移动元件的变体和家族目录,表征了复杂复合重复的类别,并定位了逆转录转导事件。该研究检测了新生转录并描绘了 CpG 甲基化谱,以定义人类转录活性逆转录元件的结构,包括着丝粒中的逆转录元件结构。这些数据扩展了对塑造人类基因组的重复区域的多样性、分布和进化的洞察力。
2022年4月1日,约翰霍普金斯大学Ariel Gershman等人在Science 在线发表题为“Epigenetic patterns in a complete human genome”的研究论文,该研究提出了对以前未解决的序列的高分辨率表观遗传研究,代表了整个近端染色体短臂、基因家族扩展和重复类别的多样化集合。该资源精确地映射了 CpG 甲基化(3228 万个 CpG)、DNA 可访问性和短读长数据集(166,058 个以前未解决的染色质免疫沉淀测序峰),以提供以前未识别或校正的基因的活性证据,并揭示临床相关的旁系同源物特异性调节。对来自六个不同个体的人类着丝粒的 CpG 甲基化进行探测,可以估计动粒定位的变异性。该分析提供了一个框架,可以用来研究人类基因组中最难以捉摸的区域,从而深入了解表观遗传调控。




当前的人类参考基因组由基因组参考联盟 (GRC) 于 2013 年发布,最近一次修补是在 2019 年 (GRCh38.p13)。该参考基因组可追溯到人类基因组计划 ,并且在过去的二十年中不断改进。与竞争的 Celera 和大多数基于“猎枪”序列组装的现代测序项目不同,GRC 组装是由通过辐射沿人类基因组排序和定向的测序细菌人工染色体 (BAC) 构建的杂交、遗传连锁和指纹图谱。然而,BAC 克隆的局限性导致重复序列的代表性不足,并且来自多个个体的 BAC 的机会性组装导致了单倍型的镶嵌。结果,几个 GRC 组装间隙由于其侧面不兼容的结构多态性而无法解决,并且许多其他重复和多态性区域未完成或组装不正确。
GRCh38 参考组装包含 151 兆碱基对 (Mbp) 的未知序列,分布在整个基因组中,包括着丝粒周围和亚端粒区域、扩增基因阵列和核糖体 DNA (rDNA) 阵列,所有这些都是基本基因组细胞过程所必需的。一些最大的参考缺口包括人类卫星(HSat)重复阵列和所有五个近端着丝粒染色体的短臂,它们在 GRCh38 中表示为未知碱基的多兆碱基延伸。尽管人类基因组计划和 GRC 都完成了提高参考质量的努力,但在随后的几年中缩小剩余差距的进展有限。
长读长鸟枪法测序克服了基于 BAC 的组装的局限性,绕过了基因组之间结构多态性的挑战。PacBio 的多千碱基单分子读取被证明能够解决 GRCh38中的复杂结构变异和间隙,而 Oxford Nanopore 的 >100-kbp “超长”读数能够完整组装人类着丝粒(染色体 Y),然后是组装整个染色体(染色体 X)。然而,这些技术的高错误率 (>5%) 对长的、几乎相同的重复阵列的组装提出了挑战。PacBio 最新的“HiFi”循环共有测序提供了 20-kbp 读取长度的折衷方案,错误率为 0.1%。
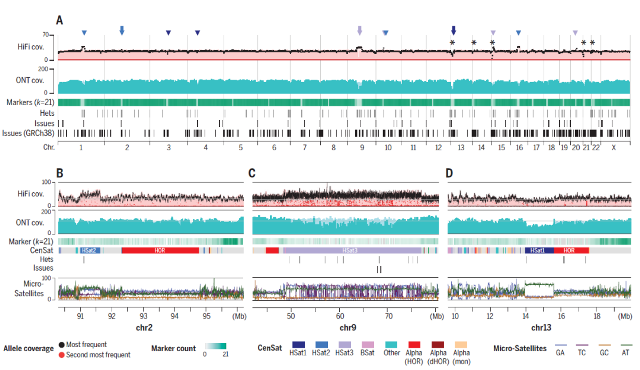
测序覆盖率和组装验证(图源自Science )
为了完成基因组的最后剩余区域,该研究利用 PacBio HiFi 和 Oxford Nanopore 超长读长测序的互补方面来组装一致的纯合 CHM13hTERT 细胞系(以下简称 CHM13)。由此产生的 T2T-CHM13 参考组装消除了一个 20 年前的屏障,该屏障隐藏了基于序列的分析的 8% 的基因组,包括所有着丝粒区域和五个人类染色体的整个短臂。在这里,该研究描述了一个真正完整的人类参考基因组的构建、验证和初步分析,并讨论了它对该领域的潜在影响。
该研究针对剩余 8% 的基因组,提供了人类基因组的完整 30.55 亿碱基对序列 T2T-CHM13,其中包括除 Y 之外的所有染色体的无间隙装配,纠正了之前的参考序列,并介绍了近 2 亿个碱基对序列,其中包含 1956 个基因预测,其中 99 个预测为蛋白质编码。 完成的区域包括所有着丝粒卫星阵列、最近的节段重复和所有五个近端着丝粒染色体的短臂。
总之,与过去 20 年的任何基因组参考版本相比,T2T-CHM13 组装增加了五个完整的染色体臂和更多的额外序列。这 8% 的基因组并没有因为缺乏重要性而被忽视,而是因为技术限制。高精度长读长测序终于消除了这一技术障碍,能够对整个人类基因组的基因组变异进行全面研究,预计这将推动人类基因组健康和疾病的未来发现。
(来源:iNature)
参考文献:
[1] Sergey Nurk, Sergey Koren, Arang Rhie, et al. The complete sequence of a human genome[J].Science. 31 Mar 2022. DOI: 10.1126/science.abj6987.
[2] Sergey Aganezov, Stephanie M. Yan, Daniela C. Soto, et al. A complete reference genome improves analysis of human genetic variation, Science, 376, 6588, (2022).
/doi/10.1126/science.abl3533
[3] Mitchell R. Vollger, Xavi Guitart, Philip C. Dishuck, et al. Segmental duplications and their variation in a complete human genome, Science, 376, 6588, (2022).doi/10.1126/science.abj6965
[4] Altemose N, Logsdon GA, Bzikadze AV, et al. Complete genomic and epigenetic maps of human centromeres. Science. 2022 Apr;376(6588):eabl4178. doi: 10.1126/science.abl4178. Epub 2022 Apr 1. PMID: 35357911.
[5] Hoyt SJ, Storer JM, Hartley GA, et al. From telomere to telomere: The transcriptional and epigenetic state of human repeat elements[J]. Science. 2022 Apr;376(6588):eabk3112. doi: 10.1126/science.abk3112. Epub 2022 Apr 1. PMID: 35357925.
[6] Gershman A, Sauria MEG, Guitart X, et al. Epigenetic patterns in a complete human genome. Science. 2022 Apr;376(6588):eabj5089. doi: 10.1126/science.abj5089. Epub 2022 Apr 1. PMID: 35357915.
链接:https://mp.weixin.qq.com/s/lbNfdVdqhSPxUCxGR6cDdg

